
Qu'est que c'est auto-immune poliglandular syndrome ¿tipo 1?
El síndrome poliglandular autoinmune tipo 1 (APS1) es una afección autoinmune que produce insuficiencias múltiples endocrine glándulas También se conoce como síndrome poliendocrino autoinmune tipo 1, poliendocrinopatía-candidiasis.ectodermique dystrophie (APECED), síndrome de Whitaker y síndrome de candidiasis-hipoparatiroidismo-enfermedad de Addison, entre sus muchos otros nombres.
APS1 fue descrita por primera vez por el Dr. Thomas Addison en el siglo XIX.
¿Quién contrae el síndrome poliglandular autoinmune tipo 1?
APS1 se hereda, y las mujeres y las niñas tienen un poco más de probabilidades de desarrollar el síndrome que los hombres y los niños. Ocurre con mayor frecuencia en poblaciones étnicas particulares debido a consanguinité o la agrupación de descendientes de un fundador familiar común. Es mas prédominant en:
- Judios iraníes (1 de cada 9000)
- Sardos (1 de cada 14.400)
- Finlandeses (1 en 25,000).
APS1 es raro en otras poblaciones.
¿Qué causa el síndrome poliglandular autoinmune tipo 1?
APS1 es causado por gène mutations en el gen regulador autoinmune, AIR, en chromosome 21q22. Se hereda en un autosomique patrón recesivo (dos copias de un gen anormal deben estar presentes para que se desarrolle el síndrome). Estas mutaciones genéticas conducen a autoanticorps y causa chronique inflammatoire cellule les Infiltrés en los órganos afectados
¿Cuáles son las características clínicas del síndrome poliglandular autoinmune tipo 1?
Los síntomas aparecen con mayor frecuencia en niños de 3 a 5 años, la mayoría de los casos de APS1 han aparecido en la adolescencia temprana, y todos los casos en el momento en que un individuo está en sus 30 años.
APS1 se basa en tres características clínicas principales:
- Mucocutáneo candidiasis que afecta la piel y muqueux membranes
- Hipoparatiroidismo, que produce entumecimiento y hormigueo en la cara y las extremidades, calambres y dolores musculares, debilidad y fatiga debido a los bajos niveles de calcio circulante.
Enfermedad de Addison, una insuficiencia de las glándulas suprarrenales, que se presenta con cambios en la piel. pigmentation, pérdida de apetito y pérdida de peso, fatiga, presión arterial baja y fatiga.
Si bien es menos común, otras características posibles de este síndrome pueden incluir:
- Hipogonadotrópico hypogonadisme
- Pernicieux anémie
- Activo crónico hépatite
- Asplenia
- Queratoconjuntivitis
- Interstitiel néphrite
Diabetes mellitus tipo 1
- Colelitiasis
- Alopécie areata
- Malabsorption
Le vitiligo
Signos cutáneos de APS tipo 1
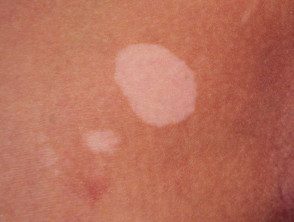
Le vitiligo
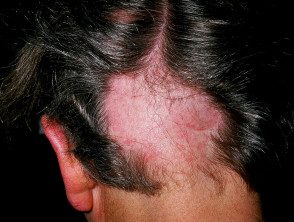
Alopécie areata

Maladie d'Addison
¿Cómo se diagnostica el síndrome poliglandular autoinmune tipo 1?
Si un individuo presenta evidencia de más de una deficiencia endocrina, se pueden realizar más pruebas para confirmar el síndrome poliglandular autoinmune tipo 1 (APS1), que incluye:
- ONGLE sérum pantalla autoinmune
- Pruebas de función del órgano final.
Los posibles análisis de sangre adicionales pueden incluir pruebas de testostéroneestradiol folliculehormona estimulante (FSH), hormona luteinizante (LH), prolactina, hormona adrenocorticotrópica (ACTH), plasma actividad de renina, niveles de electrolitos y un conteo sanguíneo completo.
Se pueden tomar hisopos y raspados de piel para detectar Candida albicans.
¿Cómo se trata el síndrome poliglandular autoinmune tipo 1?
El tratamiento de APS1 dependerá de sus características específicas.
La candidiasis mucocutánea se trata con agentes antimicóticos orales, como fluconazol o itraconazol.
- El hipoparatiroidismo se trata con una combinación de calcio oral y vitamina D (generalmente, calcitriol).
La enfermedad de Addison se trata con corticosteroides orales y mineralosteroides. Los trasplantes de glándulas suprarrenales también pueden estar indicados.
¿Cuál es el resultado del síndrome poliglandular autoinmune tipo 1?
Les prévoir para APS1 es variable. Las tasas de supervivencia han mejorado mucho desde la década de 1970.
