
La classification internationale 2017 des syndromes d'Ehlers-Danlos
Voir Ehlers-Danlos syndrome.
Des sous-types spécifiques du syndrome d'Ehlers-Danlos (EDS) ou des syndromes d'Ehlers-Danlos sont décrits ci-dessous. [1,2]. Ils comprennent:
- Syndrome d'Ehlers-Danlos classique (SEDc)
- Syndrome d'Ehlers-Danlos classique (sEDc)
- Syndrome d'Ehlers-Danlos cardio-valvulaire (SEDcv)
- Vasculaire Syndrome d'Ehlers-Danlos (SEDv)
- Syndrome d'Ehlers-Danlos hypermobile (SEDh)
- Syndrome d'Arthrochalasie d'Ehlers-Danlos (EDA)
- Syndrome de dermatosparaxis d'Ehlers-Danlos (dEDS)
- Syndrome cyphoscoliotique d'Ephlers-Danlos (SEDk)
- Fragile cornée syndrome (SBC)
- Syndrome spondylodysplasique d'Ehlers-Danlos (SEDp)
- Syndrome musculocontractural d'Ehlers-Danlos (mcEDS)
- Syndrome myopathique d'Ehlers-Danlos (SEDm)
- Syndrome parodontal d'Ehlers-Danlos (EPS)
Syndrome d'Ehlers-Danlos
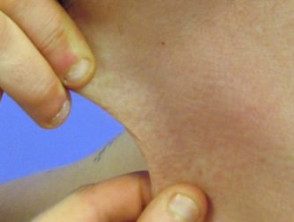
Hyperélasticité de la peau du cou.
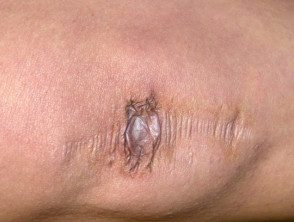
Cicatrisation extensive chez les patients atteints du syndrome d'Ehlers-Danlos.
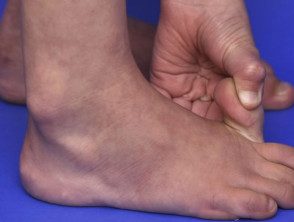
Articulations hypermobiles
Voir plus d'images du syndrome d'Ehlers-Danlos.
Syndrome d'Ehlers-Danlos classique (SEDc)
Le modèle d'héritage du cEDS est autosomique dominant.
- Les touchés les gènes dans cEDS, ils sont COL5A1, COL5A2et COL1A1 qui affecte le type V et le type I collagène.
- 50% des patients diagnostiqués avec un SEDc ont un de novo mutation (c'est-à-dire qu'aucun des parents n'est concerné).
- La gravité varie considérablement, même au sein d’une même famille.
- Vasculaire fragilité peut inclure une dissection spontanée ou une rupture d'artères de taille moyenne.
- Généralement, les patients atteints de SEDc présentent une hyperextensibilité cutanée marquée et élargie. atrophique cutané cicatrices et généralisé hypermobilité articulaire.
- Les autres fonctionnalités du cEDS sont hémosmosine déclaration (particulièrement visible sur les tibias et le dos des mains), sous-cutanée sphéroïdes (lobes graisseux calcifiés après perte de leur apport sanguin) et pseudotumeurs de mollusques (lésions/cicatrices cutanées charnues épaissies sur les genoux et les coudes).
- Il n'y a pas de stries (striations) sur le cEDS.
- Électron typique microscopie Les résultats sont des « fleurs de collagène » ou des « choux-fleurs » dus à une fibrillogenèse anormale.
- Les complications du SEDc sont les déformations des valvules cardiaques, la dilatation aortique et aortique dissection
Syndrome d'Ehlers-Danlos classique (sEDc)
Le modèle d’héritage de clEDS est autosomique récessif.
- Les touchés gène dans clEDS est TNXB qui affecte la protéine Tenascin XB.
- Les patients atteints de CLED présentent une hypermobilité articulaire généralisée, une peau hyperextensible, douce et veloutée et des ecchymoses sévères.
- Il n’y a pas de cicatrices atrophiques dans le clEDS.
Syndrome d'Ehlers-Danlos cardio-valvulaire (SEDcv)
Le mode de transmission du cvEDS est autosomique récessif.
- Le gène affecté dans le cvEDS est COL1A2 affectant le collagène protéique de type I.
- Les patients atteints de SEDcv présentent des signes légers de SED, mais présentent une anomalie aortique grave/une atteinte valvulaire cardiaque nécessitant une intervention chirurgicale.
Syndrome vasculaire d'Ehlers-Danlos (vEDS)
Le modèle d'héritage vEDS est autosomique dominant.
- Les gènes affectés dans le vEDS sont COL3A1 qui affecte le collagène de type III et COL1A1 qui affecte le collagène de type 1.
- 50% des patients diagnostiqués avec SEDv ont un de novo mutation (c'est-à-dire qu'aucun des parents n'est affecté).
- L'espérance de vie médiane est de 50 ans.
- Les caractéristiques cliniques subtiles ou absentes du SEDv chez certains patients rendent ce sous-type difficile à diagnostiquer, la mort subite dans les années 30 ou 40 étant la seule caractéristique révélatrice.
- Le SEDv peut être identifié à la naissance s’il existe des déformations notables du pied bot ou des luxations de la hanche.
- Les caractéristiques du vEDS sont des ecchymoses graves et fines translucide aspect caractéristique de la peau et du visage (joues tendues et creuses, lèvres et nez fins et yeux proéminents).
- Les complications du vEDS sont artériel dissection (y compris aorte), rupture (y compris rupture intestinale, le côlon sigmoïde étant le site le plus fréquent) et complications obstétricales (rupture utérine, post-partum massif hémorragie)
Syndrome d'Ehlers-Danlos hypermobile (SEDh)
Le mode de transmission du SEDh est autosomique dominant.
- Le gène affecté dans le hEDS est inconnu.
- Les patients atteints d’HEDS ne sont pas sujets à des complications potentiellement mortelles, mais leurs effets peuvent être débilitants.
- Les caractéristiques du SEDh sont une hypermobilité articulaire généralisée, une hypermobilité vertébrale et des luxations ou subluxations articulaires fréquentes (épaule, rotule ou articulation temporomandibulaire).
- Il n'y a pas de pseudotumeurs de mollusques.
- Les complications du SEDh sont la dilatation de la racine aortique, autonome dysfonctionnement (orthostatique postural tachycardie syndrome), sévère chronique douleur articulaire, scolioseet prématuré arthrose.
Syndrome d'Arthrochalasie d'Ehlers-Danlos (EDA)
Le modèle de transmission de l'aEDS est autosomique dominant.
- Les gènes affectés dans les DEA sont COL1A1 et COL1A2 qui affecte le collagène de type I.
- Comme le mot le décrit, arthro- signifie articulation et -chalasie signifie relaxation ; La mutation produit un collagène anormalement faible.
- Les patients atteints de DEA courent un risque à vie de concurrent luxation de plusieurs articulations majeures.
- Les caractéristiques du DAE sont hypotonie, hypermobilité articulaire généralisée sévère, congénital bilatéral luxation de la hanche et ostéopénie.
Syndrome de dermatosparaxis d'Ehlers-Danlos (dEDS)
Le mode de transmission du dEDS est autosomique récessif.
- Le gène affecté dans dEDS est ADAMTS2 affectant la protéine ADAMTS-2.
- Sparaxis est dérivé d'un mot grec signifiant « déchirer ».
- Les caractéristiques du SEDD sont une fragilité cutanée sévère, une peau relâchée et redondante, des fontanelles à fermeture retardée, des sclères bleues, viscéral fragilité et retard de croissance postnatal (c'est-à-dire petite taille).
- Il y a des hiéroglyphes caractéristiques fibrilles' dans microscope électronique de la peau
Syndrome cyphoscoliotique d'Ephlers-Danlos (SEDk)
Le mode de transmission du KEDS est autosomique récessif.
- Les gènes affectés dans dEDS sont PLOD1 qui affecte la protéine LH1 et FKBP14 qui affecte la protéine FKBP22.
- Gène mutations entraîner un déficit en lysyl hydroxylase, qui est un modificateur de collagène enzyme important pour former des liaisons croisées entre les trimères de collagène afin d’augmenter la force du collagène.
- Le KEDS se caractérise par une apparition précoce progressive Cyphoscoliose, hypotonie, retard moteur global, sclère bleue avec pression intraoculaire élevée, hypermobilité articulaire généralisée et ecchymoses faciles.
Syndrome de cornée fragile (SBC)
Le mode de transmission du BCS est autosomique récessif.
- Les gènes affectés dans dEDS sont PRDM5 qui affecte la protéine PRDM5 et ZNF469 affectant le ZNF469.
- Les patients atteints de BCS ont une cornée fine et fragile qui peut se perforer spontanément ou après une perforation mineure. traumatisme.
- Les caractéristiques du BCS sont une cornée fine, un kératocône et un bleu. sclérotique, risque de rupture du globe, de décollement de rétine et de forte myopie.
Syndrome spondylodysplasique d'Ehlers-Danlos (SEDp)
Le mode de transmission du spEDS est autosomique récessif.
- Les gènes affectés dans le spEDS sont B4GALT7 qui affecte la protéine beta4GalT7, B3GALT6 qui affecte beta3GalT6, et SLC39A13 affectant ZIP13.
- Les caractéristiques du SPEDS sont une petite taille, une hypotonie, une courbure des extrémités, une ostéopénie et une peau hyperextensible, fine et pâteuse.
Syndrome musculocontractural d'Ehlers-Danlos (mcEDS)
Le mode de transmission du mcEDS est autosomique récessif.
- Les gènes affectés dans le mcEDS sont TCSPS14 qui affecte la protéine D4ST1 et DSE affectant le DSE.
- Les caractéristiques du mcEDS sont congénitales contractures (pied équin varus), récurrent luxations, pouces en adduction dans l'enfance, peau hyperextensible, ecchymoses faciles, peau fragile et cicatrices atrophiques.
Syndrome myopathique d'Ehlers-Danlos (SEDm)
Les modes de transmission du mEDS peuvent être autosomiques dominants ou autosomiques récessifs.
- Le gène affecté dans mEDS est COL12A1 qui affecte le collagène de type XII.
- La faiblesse musculaire présente pendant la petite enfance et l’enfance a tendance à s’améliorer à l’âge adulte, avec une certaine détérioration après 40 ans.
Syndrome parodontal d'Ehlers-Danlos (EPS)
Le mode de transmission du SEDp est autosomique dominant.
- Les gènes affectés dans le pEDS sont C1R qui affecte la protéine C1r et C1S cela affecte les C1.
- Les caractéristiques du SEDp sont graves et apparaissent précocement parodontitegomme détachée prétibial assiettes, hypermobilité articulaire et peau hyperextensible.