
O que é protoporfiria eritropoiética?
A protoporfiria eritropoiética (PPE) faz parte de um grupo de genético doenças chamadas porfirias. A EPP é devida a uma deficiência hereditária de enzima ferroquelatase A atividade reduzida desta enzima causa um acúmulo da substância química protoporfirina na pele; Resultando em fotossensibilidade A pele danifica a pele. Níveis anormalmente elevados de protoporfirina raramente podem causar doença hepática.
Quem contrai protoporfiria eritropoiética?
Predomínio de EPI na Europa varia de 1 em 55.000 a 1 em 150.000 da população. Afeta igualmente homens e mulheres, e pessoas de todas as raças podem usar EPI.
Acredita-se que a PPE seja devida a uma perda composta de função mutação no gene codificação de ferroquelatase (FECH; 612386) encontrada em cromossoma 18q21. Normalmente, há uma mutação em um gene, bem como em um segundo gene de baixa expressão alelo. Tanto homens como mulheres são igualmente afetados. Raramente, há um ganho de função da mutação ALA sintase (ALAS)-2 (herança dominante ligada ao X). Portanto, a herança pode ser autossômico recessivo ou autossômico dominante com incompleto penetração (96% de pacientes no Reino Unido).
A mielodisplasia é uma neoplásico proliferação das células da medula óssea e causas cromossômico instabilidade. A PPE adquirida foi descrita em uma mulher idosa com mielodisplasia e foi considerada devido à perda por nocaute do cromossomo 18.
Quais são as características clínicas da protoporfiria eritropoiética?
Cutâneo sintomas e sinais de EPP
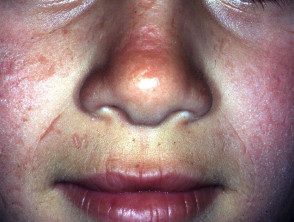
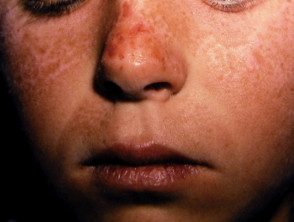
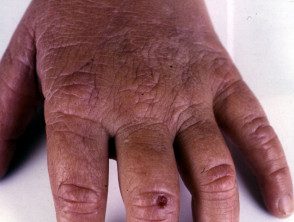
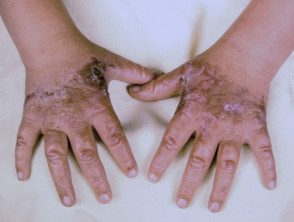
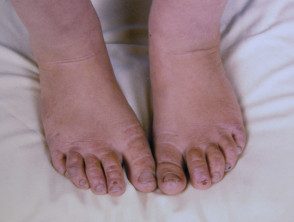
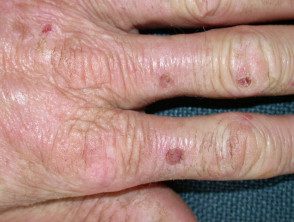
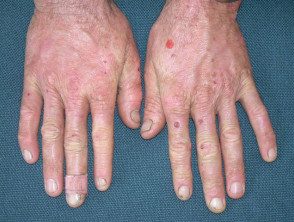
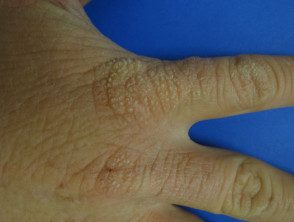
Os primeiros sintomas geralmente aparecem na primeira infância e se apresentam como uma sensação de queimação desconfortável ou dolorosa na pele após a exposição ao sol. Ocorre com mais frequência na parte superior das mãos e pés, rosto e orelhas. Na maioria dos casos, as alterações visíveis são ligeiras. A pele afetada pode ficar vermelha, inchada e com bolhas. Mais tarde, aparecem cicatrizes e, às vezes, pele muito espessa, principalmente nas bochechas, nariz e nós dos dedos das mãos.
O EPP leve causa inchaço visível e leve desconforto após a exposição ao sol. Afeta o rosto e o dorso das mãos e dos pés.
Protoporfiria eritropoiética
Protoporfiria eritropoiética
Protoporfiria eritropoiética
Protoporfiria eritropoiética
Protoporfiria eritropoiética
Protoporfiria eritropoiética
Protoporfiria eritropoiética
Protoporfiria eritropoiética
Protoporfiria eritropoiética
Doença hepática em EPP
Pessoas com doença hepática induzida por EPP geralmente apresentam alterações leves nos exames de sangue do fígado. Por volta de 10%, desenvolve-se doença hepática mais grave, apresentando desconforto, dor sob as costelas à direita, icterícia e aumento da fotossensibilidade.
Os cálculos biliares são comuns em pacientes com PPE.
Como é diagnosticada a protoporfiria eritropoiética?
O diagnóstico de PPE é frequentemente feito durante a infância.
- Pode-se observar que os glóbulos vermelhos do paciente emitem fluorescência ultravioleta. microscopia.
- O diagnóstico de PPE é confirmado pelo aumento dos níveis de protoporfirina no sangue e redução da atividade da enzima ferroquelatase. A gravidade dos achados bioquímicos é variável.
- O teste genético está disponível em alguns centros.
- UMA biópsia Também pode ser útil porque o EPP possui alguns recursos em histopatologia.
Os seguintes testes de monitoramento são frequentemente realizados de tempos em tempos.
- Hemograma completo para avaliar anemia
- Testes de ferro: a deficiência de ferro pode estar presente e deve ser monitorada
- Testes de função hepática
-
Níveis de vitamina D
Qual tratamento está disponível para a protoporfiria eritropoiética?
A fotossensibilidade ao longo da vida é o principal problema da PPE sem complicações de doença hepática.
- Evite a exposição desnecessária à luz solar e use roupas de proteção e chapéus de abas largas. Considere o escurecimento das janelas.
- Outras fontes de luz também podem causar sintomas, incluindo luzes fluorescentes e halógenas.
- Proteja a pele da exposição às luzes cirúrgicas durante um procedimento cirúrgico.
-
Os protetores solares podem ser úteis, especialmente formulações contendo óxido de zinco ou dióxido de titânio que refletem a luz visível.
-
A suplementação de vitamina D é apropriada em pacientes que evitam estritamente a exposição à luz solar.
Os ensaios de tratamento para PPE têm sido difíceis de avaliar. O tratamento eficaz deve reduzir a dor e aumentar o tempo livre de dor ao ar livre.
-
Afamelanotida, um α-melanócito hormônio estimulante administrado por subcutâneo implementação, foi relatado que fornece eficácia clínica e segurança em EPI. É aprovado em circunstâncias excepcionais pela Agência Europeia de Medicamentos para o tratamento de EPI (outubro de 2014).
-
Aumenta a fototerapia UVB de banda estreita melanina conteúdo e induz espessamento da pele, podendo reduzir a sensibilidade ao sol em alguns pacientes.
Pacientes individuais podem se beneficiar de antioxidantes orais, como beta-caroteno e N-acetilcisteína. Eles devem evitar a suplementação de ferro (a menos que haja deficiência grave de ferro), pois o ferro pode aumentar a fotossensibilidade na PPE.
- A colestiramina reduz hepático conteúdo de protoporfirina, mas não reduz a fotossensibilidade.
- Pacientes com EPP que também apresentam doença hepática necessitam de tratamento médico especializado e possivelmente transplante.
Outra condição mais grave, congênito A porfiria eritropoiética é agora curável por transfusão de células estaminais, abrindo esperança para o futuro, mas ainda não existe cura disponível para a PPE.