
¿Qué es la enfermedad de Fabry?
La enfermedad de Fabry es una enfermedad hereditaria rara. lisosomal trastorno de almacenamiento [1]. También se conoce como enfermedad de Anderson-Fabry y angioqueratoma corporis diffusum.
La enfermedad de Fabry causa grupos de pequeñas manchas rojas oscuras en la piel (angioqueratomas) Y muchos sistémico síntomas debido a la declaración de globotriaosilceramida en múltiples órganos.
Angioqueratomas en sospecha de enfermedad de Fabry
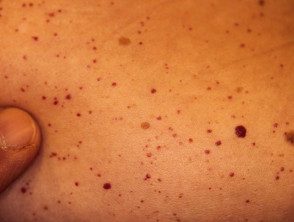
Angioqueratomas en sospecha de enfermedad de Fabry
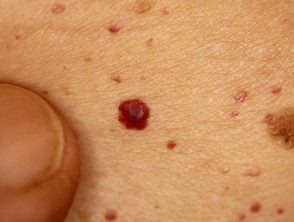
Angioqueratomas en sospecha de enfermedad de Fabry
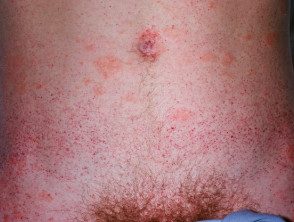
Angioqueratomas en la enfermedad de Fabry
¿Quién contrae la enfermedad de Fabry?
El estimado predominio en los hombres es aproximadamente uno de cada 40,000 a 60,000 de la población [4]. La enfermedad de Fabry generalmente afecta a los hombres más severamente y a una edad más temprana que las mujeres porque su herencia está ligada a X (los hombres solo llevan una X cromosoma mientras que las hembras tienen dos). La enfermedad de Fabry puede ocurrir en todos los grupos étnicos. [5].
La edad de inicio de la enfermedad de Fabry puede variar desde la segunda hasta la quinta década de la vida o más tarde.
La enfermedad de Fabry a menudo se presenta con síntomas inespecíficos que pueden ser leves y sutiles y comúnmente se pasa por alto o se diagnostica erróneamente, lo que lleva a una subestimación de su prevalencia. [6].
¿Qué causa la enfermedad de Fabry?
La enfermedad de Fabry es causada por un mutación de la alfa-galactosidasa A gene (GLA) mapeado en el brazo largo del cromosoma X [7]. Hay cientos de diferentes patógeno variantes de la mutación, lo que resulta en síntomas diferentes y gravedad variable [8]. Los hombres con enfermedad de Fabry transmiten el gen a todas las hijas, pero no a ninguno de sus hijos. Las mujeres con la enfermedad de Fabry tienen un 50% de posibilidades de transmitir el gen a sus hijas o hijos. Algunas mujeres se ven afectadas tan severamente como los hombres debido a la inactivación aleatoria del cromosoma X [9].
los enzima la alfa-galactosidasa A cataliza la separación (escote) de la galactosa terminal de globotriaosilceramida (Gb3). Gb3 es un intermediario en la vía degradativa del globoide (un glucosfingolípido), un componente principal de algunas membranas celulares. La falta de la enzima alfa-galactosidasa A conduce a la acumulación de Gb3 en varios tejidos, lo que conduce a la muerte celular. Los sitios de acumulación de Gb3 clínicamente más significativos son vasos sanguineos de la piel, corazón nerviosy riñones [10].
¿Cuáles son las características clínicas de la enfermedad de Fabry?
Los rasgos característicos de la enfermedad de Fabry son:
- Racimos de pequeños, rojo oscuro pápulas (angioqueratomas)
- Intolerancia al calor y sudoración reducida (hipohidrosis)
- Episodios de neuropático dolor en las manos y pies (acroparestesia), precipitado por estrés o extremos de temperatura
- Nubosidad de la córnea
- Pérdida de la audición [2]
- Síntomas abdominales como dolor, náuseas, vómitos y diarrea o estreñimiento. [3]
- Recurrente fiebre, fatiga, problemas psicológicos y sociales
- Complicaciones graves y potencialmente mortales, como daño renal, enfermedad cardíaca y accidente cerebrovascular.
Las manifestaciones dermatológicas ocurren en más del 70% de los pacientes con enfermedad de Fabry, con una edad media de inicio de 17 años. [5].
Angioqueratomas
Los angioqueratomas son vasos sanguíneos dilatados en la parte superior. dermis, presentando como pápulas rojas o negras. Las pápulas no blanquear con presión, y las lesiones más antiguas pueden convertirse verrugoso. Racimos o difuso Los angioqueratomas que aparecen por primera vez en adultos jóvenes deben alertar sobre un posible diagnóstico de enfermedad de Fabry.
En la enfermedad de Fabry, los angioqueratomas son causados por la acumulación de Gb3 en el dérmico endotelial células, lo que conduce a abultamiento e incompetencia de la pared del vaso [12]. Los angioqueratomas generalmente se localizan en el área del traje de baño (del ombligo a la parte superior de los muslos, incluidos los genitales). Más de un tercio de los pacientes también desarrollan angioqueratomas y telangiectasia en los labios y dentro de la boca [13].
No existe correlación entre la gravedad de la enfermedad y la extensión de los angioqueratomas. [11].
Hipohidrosis
La hipohidrosis es una característica común de la enfermedad de Fabry que conduce a la piel seca y a la intolerancia al calor. [11]. Es probable que sea el resultado de anormal autonómico respuesta nerviosa debido a la deposición de Gb3.
Otro cutáneo manifestaciones de la enfermedad de Fabry
La enfermedad de Fabry se identifica por otros signos cutáneos, como:
-
Linfedema de la parte inferior de las piernas. [11]
- Facial escaso y fino pelo [11]
- Fenómeno de Raynaud [14].
¿Cuáles son las complicaciones de la enfermedad de Fabry?
Las tres complicaciones más graves de la enfermedad de Fabry son la enfermedad renal, la enfermedad cardíaca y cerebrovascular enfermedad [5].
- La enfermedad renal varía desde proteinuria aislada hasta etapa terminal renal fracaso [5].
- La enfermedad cardíaca incluye ventrículo izquierdo hipertrofia, insuficiencia cardíaca, enfermedad coronaria y arritmias.
- La enfermedad cerebrovascular incluye transitoria isquémico ataques y accidentes cerebrovasculares isquémicos.
¿Cómo se diagnostica la enfermedad de Fabry?
Los pacientes masculinos y femeninos son diagnosticados de manera diferente.
- Los pacientes masculinos pueden ser diagnosticados por un análisis de sangre. Un nivel muy bajo de actividad de alfa-galactosidasa A (<3%) es suficiente para diagnosticar la enfermedad de Fabry, mientras que un nivel superior al 35% excluye la enfermedad de Fabry. Los niveles de actividad entre estas cifras requieren genético análisis para confirmar el diagnóstico.
- Las pacientes femeninas tienen actividades enzimáticas variables con una proporción significativa que muestra actividad normal, por lo tanto, el diagnóstico de la enfermedad de Fabry debe realizarse mediante análisis genético.
- Si el análisis genético no identifica una mutación que causa la enfermedad específica para la enfermedad de Fabry, un biopsia de los órganos afectados (incluida la biopsia de piel) puede considerarse; esto puede mostrar intracelular acumulación de Gb3 [5,11].
Los miembros de la familia del paciente diagnosticado también deben hacerse la prueba de la enfermedad de Fabry.
Se deben realizar análisis de orina y ECG para detectar anomalías cardíacas y renales.
Cuál es el diagnóstico diferencial para la enfermedad de Fabry?
Debido a su rareza y la amplia gama de características clínicas no específicas, la enfermedad de Fabry a menudo se diagnostica erróneamente. Puede considerarse un “gran impostor” [15].
Los angioqueratomas no son característicos de la enfermedad de Fabry, ya que pueden ocurrir lesiones solitarias en individuos sanos. También se encuentran en pacientes con hereditario hemorrágico telangiectasia y los otros trastornos lisosomales; Enfermedad de Schindler, fucosidosis, aspartilglucosaminuria, beta-manosidosis y sialidosis [11,12].
Los diagnósticos erróneos más comunes son los siguientes:
- Condiciones reumatológicas como fiebre reumática o dermatomiositis: las manifestaciones dermatológicas son diferentes en apariencia y curso temporal
- Enfermedad neuropsicológica y fibromialgia, en la que hay ausencia de afectación cardíaca, renal, nerviosa y cutánea.
- Policitemia vera y trombocitemia esencial: la enfermedad de Fabry no causa un aumento plaqueta contar
-
Telangiectasia hemorrágica hereditaria: la enfermedad de Fabry no suele causar epistaxis espontánea ni hemorragia gastrointestinal
- Intestino irritable síndrome – esto no tiene otras manifestaciones sistémicas [6,16].
¿Cuál es el tratamiento para la enfermedad de Fabry?
Una vez diagnosticados, a los pacientes varones con enfermedad de Fabry a menudo se les prescribe terapia de reemplazo enzimático, independientemente de las características clínicas. [2,11,18]. Las pacientes femeninas solo deben recibir tratamiento si tienen manifestaciones clínicas significativas.
La terapia de reemplazo de enzimas proporciona a los pacientes la enzima deficiente alfa-galactosidasa A. Reduce la gravedad y la progresión de las manifestaciones de la enfermedad, especialmente el dolor neuropático y la enfermedad renal, al reducir la deposición de Gb3 en los tejidos. Su efecto sobre las manifestaciones cardiovasculares es menos claro. [18].
Los pacientes con enfermedad de Fabry pueden requerir aportes de múltiples especialidades, incluidos médicos de cabecera, cardiólogos, nefrólogos, neurólogos y dermatólogos para controlar sus manifestaciones de órganos individuales. El dolor neuropático de la mano y los pies puede responder a anticonvulsivos, como la carbamazepina. [18,19].
El tratamiento para los angioqueratomas incluye crioterapia, electrocoagulación, cirugía escisional y láser terapia [11,12,20].
¿Cuál es el resultado de la enfermedad de Fabry?
La enfermedad de Fabry es progresivo y puede ser mortal. los pronóstico varía de paciente a paciente [21]. La supervivencia se reduce significativamente en pacientes masculinos con formas graves de enfermedad de Fabry a menos de 60 años, a menudo 40 o 50 años. La mayoría de las muertes se deben a enfermedades cardiovasculares, como accidente cerebrovascular, miocardio infartoe insuficiencia cardíaca. Muerte por insuficiencia renal, septicemia, y también se han reportado suicidios [18]. El beneficio de la terapia de reemplazo enzimático en el pronóstico general no está claro debido a la falta de evidencia actual [18,22,23].