
¿Qué es Ehlers-Danlos? síndrome?
El síndrome de Ehlers-Danlos (EDS) es el nombre dado a un grupo de trastornos hereditarios que involucran un genético defecto en colágeno y tejido conectivo síntesis y estructura [1]. Esto resulta en:
- Piel frágil e hiperelástica
- Articulaciones inestables e hiperextensibles (hipermóviles)
- Tejido frágil y vasos sanguineos.
Síndrome de Ehlers-Danlos
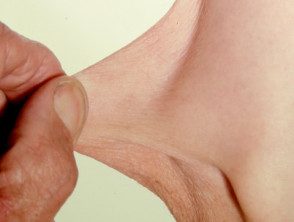
Hiperelasticidad de la piel del codo.

Hipermovilidad
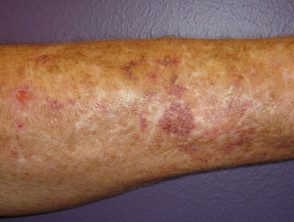
Moretones y cicatrices
¿Quién contrae el síndrome de Ehlers-Danlos?
La frecuencia global de EDS es de aproximadamente 1 en 5000. EDS puede ocurrir en hombres y mujeres de todas las razas y generalmente aparece por primera vez en adultos jóvenes.
EDS tiene autosómico dominante y autosómica recesiva Patrones de herencia.
- los dominante autosómico la enfermedad ocurre cuando solo una copia de una anormal gene Se requiere para causar una condición. Hay un 50% de posibilidades de que una descendencia herede el gen anormal de un padre afectado.
- La enfermedad autosómica recesiva requiere dos copias de un gen anormal para causar la enfermedad; por lo tanto, el niño tiene un riesgo del 25% y del 50% de desarrollar la enfermedad y ser el portador de una enfermedad, respectivamente.
Subtipos del síndrome de Ehlers-Danlos
Bajo la Clasificación Internacional 2017 para los Síndromes de Ehlers-Danlos, hay trece subtipos de SED, clasificados de acuerdo con las características clínicas, el patrón de herencia y molecular defectos genéticos.
Cada uno es un trastorno distinto que “se cumple” en una familia. Esto significa que los miembros de una familia afectada por EDS compartirán las mismas características. Algunos casos no encajan perfectamente en un tipo conocido de EDS, y un paciente puede mostrar características de más de un tipo. Las manifestaciones clínicas y su gravedad pueden variar significativamente, incluso dentro de la misma familia.
El EDS hipermóvil es el subtipo más común de EDS, seguido del EDS clásico y vascular EDS. Los otros tipos de EDS son muy raros.
Ver subtipos del síndrome de Ehlers-Danlos [view table in a new tab][2,3]
¿Qué causa el síndrome de Ehlers-Danlos?
El colágeno es uno de los principales bloques de construcción del cuerpo. Es una proteína que se encuentra ampliamente en la estructura de muchos tejidos y órganos del cuerpo. Existen varios tipos de colágeno, cada uno con diferentes propiedades. El colágeno puede proporcionar resistencia y soporte firme, puede ser elástico para permitir el movimiento, o puede usarse para unir cosas.
Un defecto genético puede causar cantidades reducidas de colágeno, desorganización del colágeno (el colágeno generalmente se organiza en paquetes) y alteraciones en el tamaño y la forma del colágeno. El subtipo de EDS depende de cómo colágeno metabolismo ha sido afectado Por ejemplo, el SED vascular es causado por una síntesis disminuida o ausente de colágeno tipo III.
Hay algunos tipos de EDS que son el resultado de otros la matriz extracelular trastornos y sus componentes (como los glicosaminoglicanos) y de defectos en intracelular Procesando.
¿Cuáles son las características clínicas del síndrome de Ehlers-Danlos?
Los signos y síntomas difieren en tipo y gravedad entre los diferentes tipos de EDS. A continuación se muestra un resumen de las características clínicas de EDS basadas en sistemas.
Rasgos faciales
- Una nariz delgada y pellizcada
- Ojos prominentes
- Orejas sin lóbulos
Afectación periodontal
- Severo e intratable inflamación del chicle
- Falta de goma adjunta
Musculoesquelético sistema
- Hipermovilidad: las articulaciones se doblan más de lo habitual. Los dedos se ven afectados con mayor frecuencia, pero todas las articulaciones pueden estar involucradas. Congénito o recurrente Las luxaciones o subluxaciones articulares pueden ocurrir particularmente en hombros, caderas y rodillas.
- Talinos equinovaro congénito (pie zambo): esta es una deformidad del desarrollo del pie donde uno o ambos pies se giran hacia adentro y hacia abajo.
- Cifoscoliosis congénita o de inicio temprano: esta es una curvatura anormal de la columna vertebral.
- Aracnodactilia: dedos de manos y pies anormalmente largos y delgados
- Congénita o leve, de inicio tardío de hipotonia y retraso motor bruto
Hipermovilidad en el síndrome de Ehlers-Danlos
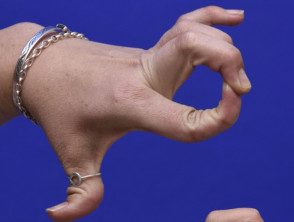
Articulaciones hipermóviles
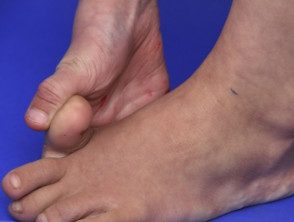
Articulaciones hipermóviles
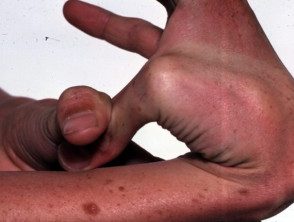
Articulaciones hipermóviles
Piel
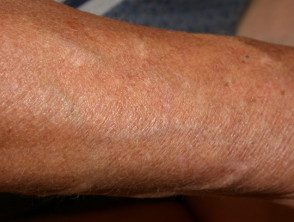
- Hiperextensibilidad de la piel: es fácil separar la piel del cuerpo y, una vez liberada, se retrae a su estado original.
- Delgado y translúcido piel con vasos sanguíneos subyacentes visibles
- Piel fragilidad – la piel se divide y se rompe fácilmente.
- Pueden aparecer hematomas y hematomas después de lesiones triviales y se pueden ver en sitios no propensos a trauma.
- Acrogeria: piel delgada y arrugada, particularmente en manos y pies
- Atrófico cutáneo cicatrices: las heridas sanan muy lentamente, lo que da como resultado grandes cicatrices en la boca de los peces o en el papel de los cigarrillos.
- Pliegues epicanticos hacer que el puente de la nariz parezca ancho.
- Pseudotumores de moluscos: estos son pequeños bultos esponjosos de 2–3 cm de diámetro sobre puntos de presión como las rodillas y los codos.
- Subcutáneo Esferoides: lóbulos grasos calcificados que son el resultado de un suministro sanguíneo insuficiente.
- Nódulos – bultos pequeños y firmes justo debajo de la superficie de la piel que aparecen con mayor frecuencia en los brazos y las espinillas.
- Pretibial placas
Hiperextensibilidad en el síndrome de Ehlers-Danlos
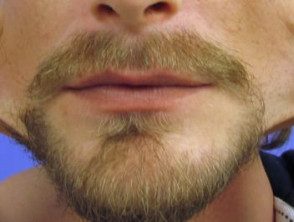
Hiperelasticidad de la piel de las mejillas.
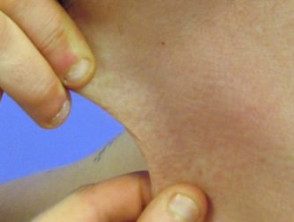
Hiperelasticidad de la piel del cuello.
Hiperelasticidad de la piel del codo.
Contusiones en el síndrome de Ehlers-Danlos
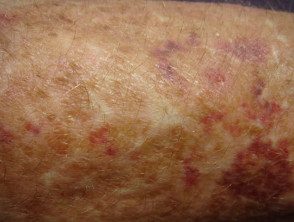
Síndrome de Ehler Danlos
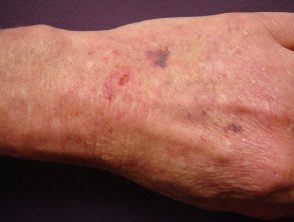
Síndrome de Ehlers Danlos
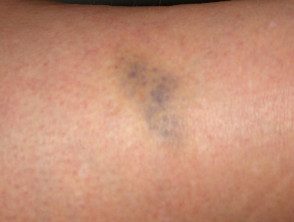
Brusing
Cicatrices atróficas en el síndrome de Ehlers-Danlos
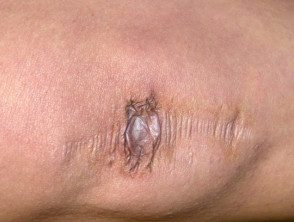
Amplia cicatrización en pacientes con síndrome de Ehlers-Danlos.
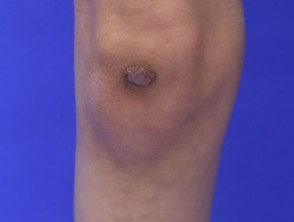
Cicatriz atrófica
Cicatrices hipopigmentadas
Síndrome de Ehlers-Danlos cutáneo
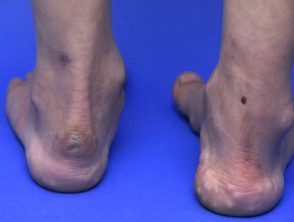
Pseudotumor en el tobillo
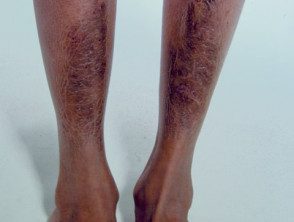
Placas pretibiales
Ver más imágenes del síndrome de Ehlers-Danlos.
Ojo
- Delgado córnea, con o sin ruptura
- Azul esclerótico
- Queratocono: la córnea tiene forma de cono
- Queratoglobo: protrusión globular y adelgazamiento de la córnea
Defectos internos de colágeno
- Soplo cardíaco (prolapso de la válvula mitral) y paredes debilitadas de intestinos, arterias y útero, que pueden romperse
- Autonómico disfunción – pérdida transitoria de la conciencia, palpitaciones, mareos, dolor en el pecho y falta de aliento
- Ortostática postural taquicardia Síndrome (POTS): latidos cardíacos rápidos, que ocurren particularmente en el cambio postural o de pie
- Neumotórax repetido: puede ser la presentación inicial de EDS.
¿Cuáles son las complicaciones del síndrome de Ehlers-Danlos?
Las posibles complicaciones del síndrome de Ehlers-Danlos incluyen:
- Crónico dolor en las articulaciones
- Mala cicatrización de heridas
- Fracaso del cierre de la herida quirúrgica
- Inicio temprano arteritis
- Ruptura prematura prematura de la membrana fetal
- Órgano hueco y ruptura vascular
- Rotura del globo, desprendimiento de retina y glaucoma
- Epilepsia [4].
¿Cómo se diagnostica el síndrome de Ehlers-Danlos?
La sospecha del síndrome de Ehlers-Danlos debe conducir a un historial y examen cuidadosos. El diagnóstico definitivo de la variante específica generalmente requiere tejido biopsia y análisis genético molecular debido a la gran variedad de defectos moleculares y la superposición clínica entre los subtipos de EDS.
Historial médico
Historia personal y familiar detallada de músculo, ocular, cutáneas, dentales, cardíacas y respiratorias.
Exámen clinico
- Control completo de la piel: evalúe la hiperextensibilidad de la piel y las cicatrices anormales.
- El beighton escala – evaluar la hipermovilidad de una articulación, en la que una puntuación de 5 o más indica generalizado hipermovilidad articular.
- El diagnóstico de subtipos de EDS se basa en criterios mayores y menores que coinciden con la presentación de signos y síntomas.
Investigaciones
Biopsia de piel para histología, fibroblastos cultura y electrón microscopía
- Al sospechar EDS cifoscoliótico, se toma una muestra de orina para medir la relación de enlaces cruzados de lisilpridinolina a hidroxilisilpiridinolina
Las pruebas genéticas moleculares confirman el diagnóstico, excepto en el caso de EDS hipermóvil, que tiene una base genética desconocida.
Cuál es el diagnóstico diferencial para el síndrome de Ehlers-Danlos?
Las enfermedades que pueden confundirse con EDS incluyen:
- Osteogénesis imperfecta
- Síndrome de Loeys-Dietz
- Displasias esqueléticas
- Mucopolisacaridosis
- Cutis síndrome de laxa
- Pseudoxantoma elástico
- Ullrich congénito muscular distrofia
- Bethlem miopatía
- Síndrome de Larsen
- Acondroplasia
- Síndrome RIN2
- síndrome de Marfan
- Condrodisplasia
- Miopatías congénitas
- Síndrome de Freeman-Sheldon.
¿Cuál es el tratamiento para el síndrome de Ehlers-Danlos?
Las pautas de manejo dependen del subtipo de EDS.
- Tomar medidas preventivas contra complicaciones graves y potencialmente mortales.
- Los pacientes deben considerar usar una pulsera MedicAlert para comunicar su estado EDS.
- Todos los pacientes con SED deben ser derivados para asesoramiento genético y especialistas apropiados.
Medidas generales
- Los niños pueden beneficiarse de almohadillas protectoras y vendajes.
- Póngase en contacto con deportes o actividades que aumentarán intracraneal Se debe evitar la presión (como tocar la trompeta) para evitar traumas indebidos.
- El paciente puede beneficiarse del ejercicio de baja resistencia y la fisioterapia.
- Considere la suplementación con calcio y vitamina D para optimizar la densidad ósea.
- Brindar apoyo psicológico luego del diagnóstico.
- Los pacientes deben usar un brazalete de advertencia médica con el “tipo de EDS” inscrito.
Medicamento
- El ácido ascórbico (vitamina C) se ha utilizado para reducir los hematomas.
Cirugía y cuidado de heridas
- La cirugía debe realizarse con precaución dado el riesgo de complicaciones vasculares.
- Las heridas deben cerrarse cuidadosamente mediante suturas sin tensión.
- Se recomiendan puntadas profundas; Deje puntos superficiales durante períodos prolongados para permitir la curación.
El embarazo
- Se recomienda un seguimiento obstétrico durante todo el embarazo debido al riesgo de trastornos hemorrágicos y vasculares, quirúrgicos y anestésico complicaciones
- Las mujeres con SED tienen un mayor riesgo de parto prematuro, posparto hemorragia, prolapso vesical y uterino, hernias abdominales y dehiscencia de la herida.
- No existen pautas de manejo obstétrico para EDS dados los amplios rangos de posibles implicaciones y la gravedad de cada subtipo; por lo tanto, los pacientes se manejan en función de cada caso.
Cribado y vigilancia
- El paciente debe tener un control regular de la presión arterial y arterial detección de enfermedades para reducir el riesgo de sangrado y complicaciones vasculares.
- No-invasor poner en pantalla – ultrasonido, tomografía computarizada (Connecticut) escaneo, ecocardiogramay imagen de resonancia magnética (Resonancia magnética) se puede realizar para identificar posibles complicaciones.
- Los procedimientos de detección invasivos deben considerarse con cuidado.
- Las exploraciones de densidad ósea de absorciometría de rayos X de energía dual (DEXA) deben realizarse cada pocos años.
- Los pacientes con kEDS deben someterse a un examen de la vista con regularidad, ya que corren el riesgo de complicaciones oculares.
¿Cuál es el resultado para los pacientes con síndrome de Ehlers-Danlos?
los pronóstico de pacientes con síndrome de Ehlers-Danlos varía ampliamente incluso dentro del mismo subtipo. La mayoría de los pacientes tienen una esperanza de vida normal y una función cognitiva. Las personas con SED vascular pueden tener una esperanza de vida más corta y tienen un mayor riesgo de muerte súbita debido a la ruptura de un órgano interno y un vaso mayor.
