
¿Qué es legius? síndrome?
El síndrome de Legius es raro genético trastorno que se describió por primera vez en 2007 [1]. También se conoce como neurofibromatosis síndrome tipo 1 [2].
El síndrome de Legius se caracteriza clásicamente por múltiples de color marrón claro. máculas, conocido como café-au-lait macules [3]. A diferencia de la neurofibromatosis tipo 1, no se encuentran tumores en el síndrome de Legius.
Café-au-lait macules
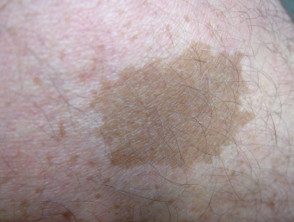
Café au lait macule
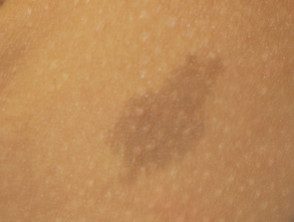
Cafe au lait macule
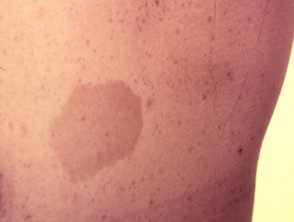
Cafe au lait macule
¿Quién contrae el síndrome de Legius?
El síndrome de Legius puede ocurrir en cualquier persona que tenga al menos un padre biológico con síndrome de Legius genéticamente confirmado. [2]. los predominio del síndrome de Legius es desconocido, pero puede ser mayor de lo esperado debido a un diagnóstico erróneo de neurofibromatosis tipo 1 [4].
¿Qué causa el síndrome de Legius?
El síndrome de Legius es una condición genética heredada en un autosómico manera dominante que implica una SPRED1 gene mutación en cromosoma 15q14. Esta mutación resulta en la función anormal de la proteína SPRED1, que es responsable de regular las rutas específicas de señalización celular involucradas en la célula. proliferación, diferenciacióny apoptosis. Ninguna otra patógeno se ha encontrado que las variantes causan el síndrome de Legius [2,3].
Como un dominante autosómico condición, el síndrome de Legius puede ocurrir en cualquier persona con al menos un padre biológico con síndrome de Legius genéticamente confirmado. En raras ocasiones, también puede ocurrir de novo, Pero estos casos no se han evaluado lo suficiente como para confirmar esto [2]. Cada niño con un padre con síndrome de Legius genéticamente confirmado tiene un 50% de riesgo de heredar la afección.
¿Cuáles son las características clínicas del síndrome de Legius?
Las características clínicas del síndrome de Legius varían significativamente en naturaleza y gravedad de una persona a otra.
Casi todos los pacientes con síndrome de Legius presentan múltiples máculas café con leche [2,3]. El número de estas máculas tiende a aumentar durante la infancia. [4].
Otro común cutáneo Las características pueden incluir:
-
Pecas en la axila y inguinal regiones
-
Lipomas [2–4].
Las características no cutáneas pueden incluir:
- Macrocefalia
- Características faciales inusuales, similares al síndrome de Noonan
- Baja estatura
- Pectus excavatum (esternón hundido) o pectus carinatum (esternón sobresaliente) [2–4].
Las características neuropsiquiátricas pueden incluir:
- Dificultades de aprendizaje
- Retraso en el desarrollo
- Trastorno por déficit de atención e hiperactividad (TDAH) [2–4].
Las discapacidades intelectuales son generalmente menos graves en pacientes con síndrome de Legius en comparación con pacientes con neurofibromatosis tipo 1 [5].
Es importante destacar que el síndrome de Legius no causa neurofibromas y Lisch nódulos, que generalmente se observan en la neurofibromatosis tipo 1 [2,3].
¿Cómo se diagnostica el síndrome de Legius?
El síndrome de Legius es difícil de diagnosticar en una base clínica sola dada su presentación clínica cutánea similar a otros trastornos con múltiples máculas café-du-lait.
Cuando se sospecha el síndrome de Legius, se pueden usar pruebas genéticas para confirmar el diagnóstico. Esto involucra:
- Análisis de secuencia de SPRED1
- Análisis de eliminación / duplicación (si el análisis de secuencia no es notable)
- Un panel de múltiples genes para SPRED1 y otra genes de interés [2,3].
Los hallazgos sospechosos que pueden justificar las pruebas genéticas para el síndrome de Legius incluyen:
-
Máculas café con leche sin otras características clínicas, lo que sugiere neurofibromatosis tipo 1
- Un padre con máculas café con leche sin características clínicas, sugiriendo nuevamente neurofibromatosis tipo 1 [2].
Cuál es el diagnóstico diferencial para el síndrome de Legius?
El síndrome de Legius a menudo se diagnostica erróneamente porque sus manifestaciones pigmentarias son muy similares a las observadas en otros síndromes con múltiples lentigos. El diagnóstico correcto es crítico para guiar el monitoreo y manejo a largo plazo.
Neurofibromatosis tipo 1
-
La neurofibromatosis tipo 1 es el diagnóstico erróneo más común debido a sus criterios diagnósticos clínicos similares. [2–4].
- Los neurofibromas, los nódulos de Lisch, las anomalías óseas y los gliomas ópticos no se presentan en el síndrome de Legius.
- Estas dos condiciones son difíciles de diferenciar en niños más pequeños, ya que los tumores característicos observados en la neurofibromatosis generalmente no se desarrollan hasta más adelante en la vida.
Síndrome de Noonan con múltiples lentigos.
-
El síndrome de Noonan con múltiples lentigos (anteriormente conocido como síndrome LEOPARD, involucra anormalidades cardiovasculares, del oído y genitales, que no ocurren en el síndrome de Legius [6].
Otros síndromes caracterizados por múltiples lentigos incluyen:
- Dominante autosómico café-au-lait macules
-
Síndrome de McCune-Albright.
¿Cuál es el tratamiento para el síndrome de Legius?
Los niños con síndrome de Legius deben someterse a pruebas de detección y evaluación periódicas por retraso en el desarrollo, deterioro cognitivo y problemas de comportamiento. [2].
El tratamiento del síndrome de Legius es principalmente de apoyo y debe centrarse en los problemas específicos del individuo afectado.
El manejo adecuado, si está indicado, puede incluir:
- Terapia física, del habla y / u ocupacional
- Terapia de modificación del comportamiento y terapia farmacológica (es decir, para el TDAH) [2].
¿Cuál es el resultado del síndrome de Legius?
Los pacientes con síndrome de Legius tienen un buen comportamiento general. pronóstico con una adecuada gestión basada en problemas.
Una persona afectada debe considerar el 50% de riesgo de transmisión genética a cada niño.
