
¿Qué es Vogt – Koyanagi – Harada? síndrome?
El síndrome de Vogt-Koyanagi-Harada (VKH) es una enfermedad multisistémica que se presenta con una combinación de oftalmologico, signos y síntomas neurológicos y dermatológicos.
- El principal hallazgo clínico definitorio de VKH es grave bilateral granulomatoso panuveitis (inflamación en todo el tracto uveal en el ojo).
- Cutáneo las características incluyen vitiligo, alopecia areata, y poliosis (blanco pelo)
- Neurológico Las características incluyen meningismo (síntomas de meningitis sin inflamación), pérdida de audición y tinnitus.
Signos cutáneos del síndrome de Vogt-Koyanagi-Harada
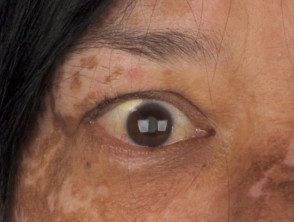
Vitiligo
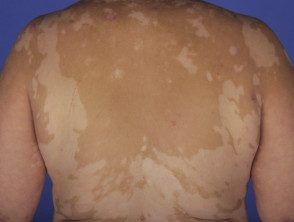
Vitiligo
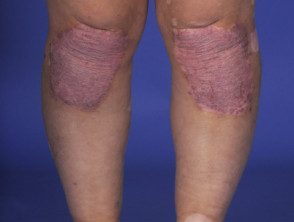
Vitiligo + psoriasis
¿Quién contrae el síndrome de Vogt-Koyanagi-Harada?
Se informa que VKH tiene una variación regional y global significativa [1]. VKH es más predominante en pacientes con piel más oscura pigmentación que en caucásicos y personas de ascendencia turca. Los pacientes con VKH son más comúnmente de origen asiático, hispano, indio, nativo americano o mediterráneo. [2]. Hay dos veces más mujeres afectadas por VKH que hombres [3].
La VKH se diagnostica principalmente en pacientes adultos y de mediana edad de 20 a 50 años, aunque también se ha informado su aparición en la infancia o la vejez. [4–6].
¿Qué causa el síndrome de Vogt-Koyanagi-Harada?
La causa exacta de VKH aún no se ha establecido. Es un autoinmune inflamatorio condición mediada por CD4 + Células T focalización melanocitos [7,8]. Genético predisposición y / o una exposición no especificada a un virus puede estar involucrada [1].
¿Cuáles son las características clínicas del síndrome de Vogt-Koyanagi-Harada?
Clásicamente, VKH tiene cuatro etapas clínicas. Las características clínicas varían, dependiendo del grupo étnico. Las características dermatológicas se observan con mayor frecuencia en el crónico etapa convaleciente [1].
los prodrómico escenario
La etapa prodrómica de VKH puede imitar viral infección y dura de 1 a 2 semanas [2]. Los síntomas son principalmente neurológicos e incluyen:
- Dolor de cabeza
- Rigidez en el cuello
- Fotofobia
- Fiebre
- Dolor orbital
- Cuero cabelludo y sensibilidad general de la piel.
- Focal déficits neurológicos (p. ej., parálisis del nervio craneal).
Los síntomas auditivos incluyen:
- Pérdida de audición (particularmente para frecuencias más altas)
- Molestias cuando se expone a sonidos a volumen normal
- Vértigo
- Tinnitus [2].
los agudo escenario
La etapa aguda de VKH afecta el ojo y dura varias semanas. [2].
- El principal síntoma de la etapa aguda de VKH es visión borrosa en ambos ojos [1].
- Los hallazgos oftalmológicos son panuveitis o posterior uveítis, con multifocal seroso desprendimientos de retina [2].
- El reconocimiento temprano y el tratamiento pueden prevenir la progresión a convalecientes crónicos y crónicos recurrente etapas de la enfermedad [7].
La etapa de convalecencia crónica
Muchos pacientes con VKH aguda pasan a la etapa de convalecencia crónica de VKH, aproximadamente 3 a 4 meses después del inicio de la enfermedad. [9,10]. Esto generalmente dura algunos meses o años. [1,2].
- Los hallazgos oftalmológicos de la fase convaleciente crónica de VKH incluyen panuveítis no granulomatosa y despigmentación de la coroides (capa media del ojo), con un color naranja brillante fondo de ojo (un ‘resplandor del atardecer’) [10,11].
- Los hallazgos dermatológicos son poliosis de las cejas y pestañas, vitiligo y alopecia areata. [1].
La etapa recurrente crónica.
El 25% de los pacientes con VKH aguda desarrollan la fase recurrente crónica de la enfermedad 6–9 meses después de la presentación inicial [1,2].
- Los hallazgos oftalmológicos son granulomatosos recurrentes. anterior uveítis y engrosamiento coroideo.
-
El vitiligo y la alopecia areata pueden tener fases de recaída y remisión.
¿Cuáles son los hallazgos dermatológicos en el síndrome de Vogt-Koyanagi-Harada?
Alrededor del 30% de los pacientes con VKH desarrollan hallazgos dermatológicos. [12]. El vitiligo, la poliosis y la alopecia areata generalmente ocurren en la etapa de convalecencia crónica de la enfermedad. [13].
¿Cuáles son las complicaciones del síndrome de Vogt-Koyanagi-Harada?
Los pacientes con VKH pueden desarrollar complicaciones que amenazan o perjudican la visión, como:
- Desprendimiento de retina en la fase aguda [2]
- Secundario glaucoma en la fase crónica (45% de los casos de VKH)
- Cataratas (42% de los casos [11,14,15])
- Subretinal fibrosis (40% de los casos [16])
¿Cómo se diagnostica el síndrome de Vogt-Koyanagi-Harada?
Los criterios de diagnóstico para VKH incluyen inflamación de ambos ojos sin evidencia de otro ocular enfermedad, una historia de traumao cirugía ocular [17].
Hay tres categorías distintas de la enfermedad. [17]:
- VKH completa: donde hay compromiso ocular bilateral (con criterios detallados según la etapa de la enfermedad), además de signos neurológicos y dermatológicos; Los signos neurológicos pueden resolverse, mientras que los signos dermatológicos no preceden al inicio de la uveítis.
- VKH incompleta: donde los hallazgos oftalmológicos son los de pacientes con VKH completa, más signos neurológicos o dermatológicos.
- Probable VKH: donde los hallazgos oftalmológicos son como para pacientes con VKH completa, pero sin signos neurológicos o dermatológicos.
Cuál es el diagnóstico diferencial para el síndrome de Vogt – Koyanagi – Harada?
Los diagnósticos diferenciales para los signos y síntomas oftalmológicos de la enfermedad incluyen:
- Trauma previo – incluyendo oftalmia simpática (inflamación de la conjuntiva o el globo ocular; La oftalmia simpática puede tener un cuadro clínico casi idéntico al de VKH, incluidas sus manifestaciones dermatológicas. [18]
- Infección – incluyendo bacteriano o infección micótica, tuberculosis o sífilis [1]
- Malignidad – como intraocular linfoma, sistémico linfoma o leucemia [1]
- Enfermedad inflamatoria – incluyendo posterior bilateral escleritis, sarcoidosis o coroidopatía por lupus (degradación del recubrimiento ocular debido al lupus) [1].
¿Cuál es el tratamiento para el síndrome de Vogt-Koyanagi-Harada?
La VKH aguda debe tratarse de forma agresiva con un corticosteroide sistémico (la duración mínima del tratamiento es de 6 meses), combinada con un agente ahorrador de corticosteroides, como ciclosporina, metotrexato o micofenolato mofetilo. [1]. Anti-tumor necrosis Se pueden considerar agentes de factor alfa (TNF) u otros agentes biológicos en pacientes que no responden a los corticosteroides combinados con agentes ahorradores de corticosteroides [1].
El objetivo del tratamiento es amortiguar la respuesta inflamatoria y prevenir complicaciones oculares, como el fondo de luz del atardecer, cataratas, glaucoma y neovascularización coroidea. [1].
¿Cuál es el resultado del síndrome de Vogt-Koyanagi-Harada?
Se describen mejores resultados visuales en aquellos pacientes que se someten a un tratamiento rápido y agresivo de VKH. Sin embargo, una proporción significativa de pacientes aún desarrolla complicaciones que afectan la visión. Las cataratas y el glaucoma a menudo requieren tratamiento quirúrgico [1,2].
Favorable pronóstico Los factores incluyen una buena agudeza visual inicial y el inicio del tratamiento en la etapa aguda y no crónica de la enfermedad. [11,15,19].