
Qu'est-ce que Crouzon? syndrome?
Le syndrome de Crouzon se caractérise par une variété de symptômes développementaux et craniofaciaux.
C'est un héréditaire état hérité dans un autosomique modèle dominant (une anomalie gène d'un parent peut provoquer le syndrome). Elle est également connue sous le nom de maladie de Crouzon, dysostose craniofaciale, craniosténose, syndrome d'Apert-Crouzon, acrocephalosyndactylie de type II, céphalosinodiactylie de Vogt et trigorinophalangienne. dysplasie.
Il a été décrit pour la première fois par Crouzon en 1912.
Qui contracte le syndrome de Crouzon?
Le syndrome de Crouzon est rare, bien qu'il soit toujours le syndrome de craniosynostose le plus courant (où les articulations fibreuses du crâne se ferment prématurément pendant l'enfance).
- Elle affecte 1 naissance sur 60 000.
- Il semble être également diagnostiqué chez les personnes de toutes races et ethnies.
- Il est souvent diagnostiqué à la naissance ou dans l'enfance en raison de caractéristiques faciales distinctives.
Quelles sont les causes du syndrome de Crouzon?
Le syndrome de Crouzon est généralement causé par mutations dans le fibroblastes facteur de croissance destinataire 2 (FGFR2) gén. Les FGFR3 le gène peut également être impliqué.
- Est mutation il conduit à des signaux aux cellules immatures pour devenir des cellules osseuses lors de l'embryogenèse.
- Des antécédents familiaux de syndrome de Crouzon sont présents dans les cas 50%.
- Dans les autres cas 50%, le syndrome est sporadique, du fait de nouvelles mutations génétiques.
Génétique du syndrome de Crouzon *
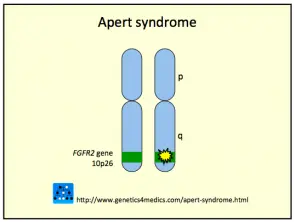
Syndrome de Crouzon2
* Image gracieuseté de Genetics 4 Medics
Quelles sont les caractéristiques cliniques du syndrome de Crouzon?
Les caractéristiques cliniques du syndrome de Crouzon varient considérablement et varient de légères à graves. La caractéristique clé est la fermeture prématurée de la calotte crânienne et des sutures de la base crânienne, et la craniosynostose.
Caractéristiques faciales
Les caractéristiques faciales distinctives associées incluent:
- Exophtalmie (protrusion anormale du globe oculaire)
- Hypertélorisme (largeur excessive entre les yeux)
- Maxillaire hypoplastique (une mâchoire sous-développée)
- Prognathisme mandibulaire (protrusion de la mâchoire inférieure)
- Une lèvre supérieure courte
- Un nez pointu.
Défauts visuels
Les défauts visuels associés au syndrome de Crouzon comprennent:
- Amblyopie (obscurité de la vue sans changement apparent dans les structures oculaires)
- Amétropie (erreur réfractaire)
- Strabisme (incapacité à atteindre binoculaire vision due à des déséquilibres musculaires dans les globes oculaires).
Ces défauts visuels sont dus à des lésions cornéennes, cascades (opacification du cristallin de l'œil ou de la membrane transparente qui l'entoure), optique atrophie (détérioration du nerf optique) et colobome de l'iris (un trou dans l'iris).
Autres caractéristiques
Les autres caractéristiques associées au syndrome de Crouzon comprennent:
- Fonction mentale diminuée et risque accru intracrânien pression et crises
- Symptômes respiratoires dus au rétrécissement de la nasopharyngé étape et dévié pulpe ou d'autres anomalies structurelles
- Perte d'audition et / ou Ménière maladie (trouble de l'oreille interne caractérisé par des épisodes de vertige, bourdonnements dans les oreilles, perte auditive et pression dans l'oreille)
- Anomalies squelettiques, notamment fusion de la colonne vertébrale.
Les principaux dermatologiques signer du syndrome de Crouzon est acanthose nigricans, dans lequel il y a épaissi, hyperpigmenté peau avec une sensation veloutée qui affecte le cou, le torse et le visage. Il apparaît généralement au début de la puberté.
Acanthosis nigricans *
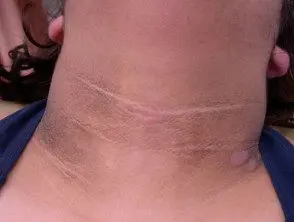
Acanthosis nigricans
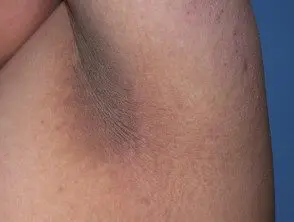
Acanthosis nigricans
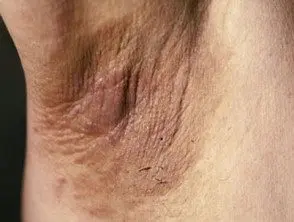
Acanthosis nigricans
Comment diagnostique-t-on le syndrome de Crouzon?
Crouzon est diagnostiqué en toute confiance chez un enfant atteint de craniosynostose lorsque des mutations FGFR2 Les gènes sont détectés. Les les gènes Les parents de l'individu affecté peuvent également être testés pour le mosaïcisme (qui a des cellules de deux types génétiquement différents ou plus).
Comment traite-t-on le syndrome de Crouzon?
Le traitement standard du syndrome de Crouzon comprend une craniectomie précoce (ablation chirurgicale d'une partie du crâne) et une reconstruction esthétique pour aider à promouvoir une croissance faciale normale.
Les soins multidisciplinaires peuvent inclure l'évaluation médicale et chirurgicale et la gestion des symptômes. Cela peut inclure:
- Ophtalmologique traitement de l'amblyopie, de l'amétropie et du strabisme
- Traitement audiologique et myringotomie pour perte auditive.
- Orientation pour traiter la pression intracrânienne en raison de hydrocéphalie (augmentation du liquide céphalorachidien dans la cavité crânienne)
- Trachéotomie (a incision dans la trachée) pour traiter l'obstruction des voies respiratoires
- Chirurgie orthodontique pour traiter les irrégularités des dents et des mâchoires.
Quel est le pronostic du syndrome de Crouzon?
L'amélioration des techniques chirurgicales, en particulier de la chirurgie craniofaciale, a considérablement augmenté la qualité de vie, les capacités intellectuelles et physiques et l'acceptation sociale des enfants atteints du syndrome de Crouzon.
La durée de vie des personnes atteintes du syndrome de Crouzon est généralement normale, mais la mortalité peut survenir en raison d'une obstruction des voies respiratoires, aigu détresse respiratoire ou augmentation de la pression intracrânienne.
