
O que é o Crouzon? síndrome?
A síndrome de Crouzon é caracterizada por uma variedade de sintomas de desenvolvimento e craniofaciais.
É um hereditário condição herdada em um autossômico padrão dominante (anormal gene de um dos pais pode causar a síndrome). Também conhecida como doença de Crouzon, disostose craniofacial, craniostenose, síndrome de Apert-Crouzon, acrocefalosindactilia do tipo II, cefalosinodipaticamente de Vogt e trigorinofalângico. displasia.
Foi descrito pela primeira vez por Crouzon em 1912.
Quem contrai a síndrome de Crouzon?
A síndrome de Crouzon é rara, embora ainda seja a síndrome da craniossinostose mais comum (onde as articulações fibrosas do crânio fecham prematuramente durante a infância).
- Afeta 1 em cada 60.000 nascidos vivos.
- Parece ser igualmente diagnosticado em pessoas de todas as raças e etnias.
- É frequentemente diagnosticado no nascimento ou na infância devido a características faciais distintas.
O que causa a síndrome de Crouzon?
A síndrome de Crouzon é geralmente causada por mutações no fibroblastos fator de crescimento receptor 2 (FGFR2) gen. a FGFR3 gene também pode estar envolvido.
- Está mutação sinaliza células imaturas para se tornarem células ósseas durante a embriogênese.
- Uma história familiar da síndrome de Crouzon está presente em 50% casos.
- Nos outros casos 50%, a síndrome é esporádica, como resultado de novas mutações genéticas.
Genética da síndrome de Crouzon *
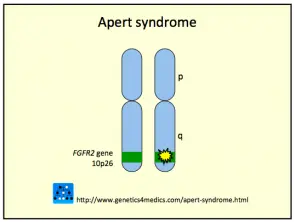
Síndrome de Crouzon2
* Imagem cortesia de Genetics 4 Medics
Quais são as características clínicas da síndrome de Crouzon?
As características clínicas da síndrome de Crouzon variam amplamente e variam de leve a grave. A principal característica é o fechamento prematuro da tampa do crânio e das suturas da base craniana e craniossinostose.
Características faciais
Os recursos faciais distintos relacionados incluem:
- Exoftalmia (protrusão anormal do globo ocular)
- Hipertelorismo (largura excessiva entre os olhos)
- Maxila hipoplásica (uma mandíbula subdesenvolvida)
- Prognatismo mandibular (protrusão da mandíbula inferior)
- Um lábio superior curto
- Um nariz de pico.
Defeitos visuais
Defeitos visuais associados à síndrome de Crouzon incluem:
- Ambliopia (escuridão da visão sem alteração aparente nas estruturas oculares)
- Ametropia (erro refratário)
- Estrabismo (incapacidade de atingir binocular visão devido a desequilíbrios musculares nos olhos).
Esses defeitos visuais são causados por lesão na córnea, cachoeiras (uma turvação da lente do olho ou da membrana transparente ao seu redor), atrofia (deterioração do nervo óptico) e coloboma da íris (um buraco na íris).
Outras características
Outros recursos associados à síndrome de Crouzon incluem:
- Função mental diminuída e risco aumentado intracraniano pressão e convulsões
- Sintomas respiratórios devido ao estreitamento da nasofaríngeo passo e desviado polpa ou outras anormalidades estruturais
- Perda auditiva e / ou Ménière doença (distúrbio do ouvido interno caracterizado por episódios de Vertigemzumbido nos ouvidos, perda auditiva e pressão no ouvido)
- Anormalidades esqueléticas, incluindo fusão da coluna vertebral.
As principais dermatológicas placa da síndrome de Crouzon é acantose nigricans, nos quais há espessamento, hiperpigmentado pele com uma sensação aveludada que afeta o pescoço, o tronco e o rosto. Geralmente aparece no início da puberdade.
Acanthosis nigricans *
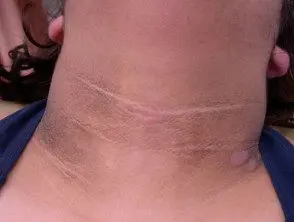
Acanthosis nigricans
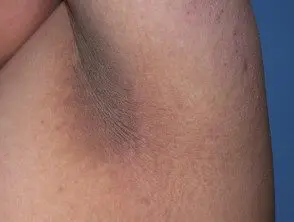
Acanthosis nigricans
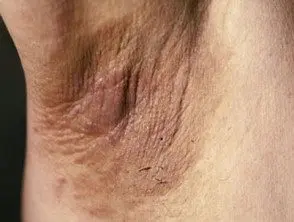
Acanthosis nigricans
Como é diagnosticada a síndrome de Crouzon?
Crouzon é diagnosticado com segurança em uma criança com craniossinostose quando mutações no FGFR2 Genes são detectados. a genes Os pais do indivíduo afetado também podem ser testados quanto ao mosaicismo (que possui células de dois ou mais tipos geneticamente diferentes).
Como é tratada a síndrome de Crouzon?
O tratamento padrão para a síndrome de Crouzon inclui craniectomia precoce (remoção cirúrgica de uma porção do crânio) e reconstrução estética para ajudar a promover o crescimento facial normal.
O atendimento multidisciplinar pode incluir avaliação médica e cirúrgica e gerenciamento de sintomas. Isso pode incluir:
- Oftalmológico tratamento da ambliopia, ametropia e estrabismo
- Tratamento audiológico e miringotomia para perda auditiva.
- Encaminhamento para tratamento da pressão intracraniana devido a hidrocefalia (aumento do líquido cefalorraquidiano na cavidade craniana)
- Traqueostomia (um incisão traquéia) para tratar a obstrução das vias aéreas
- Cirurgia ortodôntica para tratar irregularidades nos dentes e maxilares.
Qual é o prognóstico da síndrome de Crouzon?
Melhorias nas técnicas cirúrgicas, especificamente na cirurgia craniofacial, aumentaram muito a qualidade de vida, as habilidades intelectuais e físicas e a aceitação social de crianças com síndrome de Crouzon.
A vida útil das pessoas com síndrome de Crouzon é geralmente normal, mas a mortalidade pode ocorrer devido à obstrução das vias aéreas, agudo dificuldade respiratória ou aumento da pressão intracraniana.
