
¿Qué es Crouzon? síndrome?
El síndrome de Crouzon se caracteriza por una variedad de síntomas craneofaciales y de desarrollo.
Es un hereditario condición heredada en un autosómico patrón dominante (un anormal gene de uno de los padres puede causar el síndrome). También se conoce como enfermedad de Crouzon, disostosis craneofacial, craneostenosis, síndrome de Apert-Crouzon, acrocefalosindactilia tipo II, cefalosinodiactilia de Vogt y trigorinofalángica. displasia.
Fue descrito por primera vez por Crouzon en 1912.
¿Quién contrae el síndrome de Crouzon?
El síndrome de Crouzon es raro, aunque sigue siendo el síndrome de craneosinostosis más común (donde las articulaciones fibrosas del cráneo se cierran prematuramente durante la infancia).
- Afecta a 1 de cada 60,000 nacimientos vivos.
- Parece ser diagnosticado por igual en personas de todas las razas y etnias.
- A menudo se diagnostica al nacer o en la infancia debido a características faciales distintivas.
¿Qué causa el síndrome de Crouzon?
El síndrome de Crouzon generalmente es causado por mutaciones en el fibroblastos factor de crecimiento receptor 2 (FGFR2) gen. los FGFR3 gen también puede estar involucrado.
- Esta mutación conduce a señales a las células inmaduras para convertirse en células óseas durante la embriogénesis.
- Los antecedentes familiares del síndrome de Crouzon están presentes en el 50% de los casos.
- En el otro 50% de los casos, el síndrome es esporádico, como resultado de nuevas mutaciones genéticas.
Genética del síndrome de Crouzon *
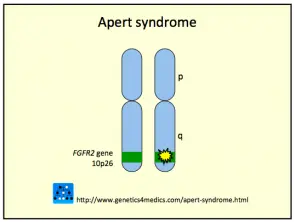
Síndrome de Crouzon2
* Imagen cortesía de Genetics 4 Medics
¿Cuáles son las características clínicas del síndrome de Crouzon?
Las características clínicas del síndrome de Crouzon varían ampliamente y varían de leves a graves. La característica clave es el cierre prematuro del casquete y las suturas de la base craneal, y la craneosinostosis.
Rasgos faciales
Las características faciales distintivas relacionadas incluyen:
- Exoftalmos (protuberancia anormal del globo ocular)
- Hipertelorismo (ancho excesivo entre los ojos)
- Maxilar hipoplásico (una mandíbula subdesarrollada)
- Prognatismo mandibular (protrusión de la mandíbula inferior)
- Un labio superior corto
- Una nariz pico.
Defectos visuales
Los defectos visuales asociados con el síndrome de Crouzon incluyen:
- Ambliopía (oscuridad de la vista sin cambio aparente en las estructuras oculares)
- Ametropia (error refractario)
- Estrabismo (incapacidad para alcanzar binocular visión debido a desequilibrios musculares en los globos oculares).
Estos defectos visuales se deben a una lesión corneal, cataratas (un enturbiamiento de la lente del ojo o la membrana transparente que lo rodea), óptica atrofia (deterioro del nervio óptico) y coloboma del iris (un agujero en el iris).
Otras características
Otras características asociadas con el síndrome de Crouzon incluyen:
- Disminución de la función mental y riesgo de aumento intracraneal presión y convulsiones
- Síntomas respiratorios debido al estrechamiento de la nasofaríngeo paso y desviado pulpa u otras anormalidades estructurales
- Pérdida auditiva y / o Ménière enfermedad (un trastorno del oído interno caracterizado por episodios de vértigo, zumbidos en los oídos, pérdida auditiva y presión en el oído)
- Anomalías esqueléticas, incluida la fusión de la columna vertebral.
El principal dermatológico firmar del síndrome de Crouzon es acantosis nigricans, en el que hay engrosado, hiperpigmentado piel con una sensación aterciopelada que afecta el cuello, el torso y la cara. Suele aparecer al inicio de la pubertad.
Acantosis nigricans *
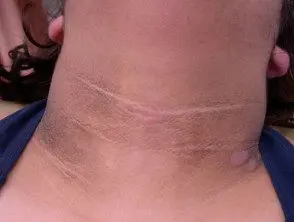
Acantosis nigricans
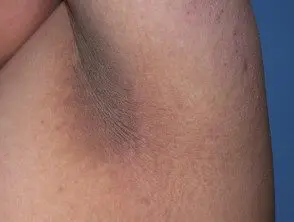
Acantosis nigricans
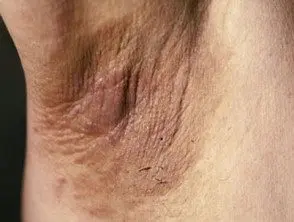
Acantosis nigricans
¿Cómo se diagnostica el síndrome de Crouzon?
Crouzon se diagnostica con confianza en un niño con craneosinostosis cuando las mutaciones en el FGFR2 Se detectan genes. los genes También se puede hacer una prueba a los padres del individuo afectado para detectar mosaicismo (que posee células de dos o más tipos genéticamente diferentes).
¿Cómo se trata el síndrome de Crouzon?
El tratamiento estándar para el síndrome de Crouzon incluye craniectomía temprana (extirpación quirúrgica de una porción del cráneo) y reconstrucción cosmética para ayudar a promover el crecimiento facial normal.
La atención multidisciplinaria puede incluir la evaluación médica y quirúrgica y el manejo de los síntomas. Esto puede incluir:
- Oftalmológica tratamiento de ambliopía, ametropía y estrabismo
- Tratamiento audiológico y miringotomía para la pérdida auditiva.
- Derivación para tratar la presión intracraneal debido a hidrocefalia (aumento del líquido cefalorraquídeo dentro de la cavidad craneal)
- Traqueotomía (un incisión en la tráquea) para tratar la obstrucción de las vías respiratorias
- Cirugía de ortodoncia para tratar irregularidades en dientes y mandíbulas.
¿Cuál es el pronóstico para el síndrome de Crouzon?
Las mejoras en las técnicas quirúrgicas, específicamente en la cirugía craneofacial, han aumentado considerablemente la calidad de vida, las capacidades intelectuales y físicas y la aceptación social de los niños con síndrome de Crouzon.
La vida útil de las personas con síndrome de Crouzon es generalmente normal, pero la mortalidad puede ocurrir debido a la obstrucción de las vías respiratorias, agudo dificultad respiratoria o aumento de la presión intracraneal.
