
What is it congenital adrenal hyperplasia?
El término hiperplasia suprarrenal congénita se refiere a las glándulas suprarrenales agrandadas. Se debe a heredado enzyme deficiencia. La hiperplasia suprarrenal congénita es el trastorno suprarrenal más común de la infancia y la niñez.
La hiperplasia suprarrenal congénita resulta de un exceso androgens (hormonas masculinas). También hay una forma severa de pérdida de sal de la condición.
¿Cómo surge la hiperplasia suprarrenal congénita?
Una deficiencia enzimática puede provocar que las glándulas suprarrenales no produzcan cantidades normales de cortisol. La disminución en el nivel de cortisol estimula la pituitary gland para liberar la hormona corticotrófica suprarrenal (ACTH). La ACTH hace que la glándula suprarrenal se agrande y produzca más cortisol, mineralocorticoides y andrógenos.
- El cortisol es un glucocorticoids. Glucocorticoides tener múltiples acciones en muchos sistemas de órganos. Aumentan el hígado glucose salida, causa atrophy de fibras musculares tipo II, inhibit formación ósea, reduce la absorción de calcio en el intestino, aumenta el gasto cardíaco, aumenta la presión arterial y aumenta la maduración pulmonar. También causan insulin resistencia, modula el eje tiroideo y reduce el número de circulantes lymphocyte cells.
- El principal mineralocorticoide es la aldosterona. Esto actúa sobre los riñones para reabsorber sodio y agua en el torrente sanguíneo y secretar potasio en la orina.
- Los andrógenos suprarrenales son dehidroepiandrosterona (DHEA), androstenediona y 11-hidroxiandrostenediona. Estos son andrógenos débiles y se convierten en los más potentes. androgynous testosterone en otros tejidos, incluida la piel. Los andrógenos regulan la secreción de hormonas sexuales por el hypothalamic-pituitario (gonadotropinas) y regulan la formación de características masculinas durante la pubertad.
La hiperplasia suprarrenal congénita puede contrastarse con Cushings Syndrome, en el que hay exceso de cortisol. Cuando esto se debe al exceso de ACTH, las glándulas suprarrenales también se agrandan y producen más andrógenos suprarrenales.
Genética de la hiperplasia suprarrenal congénita *
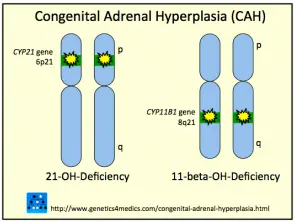
Hiperplasia suprarrenal congénita CAH
* Image courtesy of Genetics 4 Medics
Than enzymes ¿estan involucrados?
La hiperplasia suprarrenal congénita puede deberse a:
- 21-hydroxylase deficiency
- Deficiencia de 17-hidroxilasa
- Deficiencia de 3β-hidroxiesteroide deshidrogenasa
- Deficiencias parciales de enzimas.
Los signos clínicos de la hiperplasia suprarrenal congénita dependen de qué enzima carece y en qué medida.
21 deficiencia de hidroxilasa
La deficiencia de 21-hidroxilasa o hiperplasia congénita clásica representa la mayoría de los casos (95%). Puede causar problemas de salud en el período del recién nacido o no hasta la pubertad o más tarde. La deficiencia enzimática reduce el cortisol, aumenta los andrógenos y, en un tercio de los casos, reduce la producción de aldosterona.
La deficiencia de 21-hidroxilasa puede presentarse en las primeras dos semanas de vida. Puede presentarse como “pérdida de sal” acute insuficiencia suprarrenal o como ambigüedad genital en mujeres. La deficiencia parcial de la enzima 21-hidroxilasa es menos grave. Es más probable que se presente en la infancia o adolescencia tardía con signos de exceso de andrógenos.
| Características clínicas de la deficiencia de 21-hidroxilasa | |
|---|---|
| Insuficiencia suprarrenal aguda |
|
| Ambigüedad genital |
|
| Inicio tardío en mujeres |
|
| Inicio tardío en varones |
|
How was the diagnosis made?
El diagnóstico debe considerarse en los recién nacidos que presentan insuficiencia suprarrenal aguda, con virilización (mujeres) o pubertad prematura (hombres).
Las pruebas iniciales en insuficiencia suprarrenal aguda revelan un estado de pérdida de sal.
- Bajos niveles de sodio (hiponatremia)
- Altos niveles de potasio (hipercalemia)
- Bass serum aldosterona
- Cortisol sérico bajo
- High plasma renina
La deficiencia de 21-hidroxilasa se diagnostica al encontrar altos niveles de las siguientes hormonas en sangre u orina:
- Suero 17-hidroxiprogesterona
- Sulfato de plasma DHEA
- Pregnanetriol en orina
- 17-cetosteroides en orina
Una suprarrenal ultrasound Se puede realizar una exploración si los genitales del bebé parecen anormales al nacer.
Genetic Las pruebas pueden estar disponibles para identificar la genética específica mutation.
Prenatal diagnóstico
Se pueden considerar pruebas genéticas durante el embarazo si se sabe que un bebé nonato está en riesgo porque un hermano está afectado o si se sabe que ambos padres son portadores de lo anormal. gene. Las pruebas genéticas realizadas durante el embarazo incluyen:
- Muestreo de vellosidades coriónicas en la octava semana
- Amniocentesis a las 12 semanas
El diagnóstico prenatal también se puede hacer mediante el aumento de los niveles de 17-hidroxiprogesterona en amniotic líquido a las 14 semanas de gestación.
Tratamiento de la hiperplasia congénita clásica.
El tratamiento de la hiperplasia congénita clásica tiene como objetivo reemplazar el glucocorticoide (cortisol) para prevenir la secreción excesiva de ACTH, usando una pequeña dosis de dexametasona, generalmente 0,5 mg por la noche.
Los niveles de andrógenos circulantes pueden reducirse mediante el tratamiento antiandrogénico. Los medicamentos disponibles incluyen:
- Cyproterone Acetate
- Spironolactone
- Flutamide
- Finasteride
En la forma de hiperplasia suprarrenal congénita con pérdida de sal, se administra mineralocorticoide, generalmente fludrocortisona a una dosis de 0.1 mg, para mantener el volumen normal de líquido extracelular y los niveles de electrolitos. La presión arterial, los electrolitos y la actividad de la renina plasmática se controlan para evaluar la respuesta.
Deficiencia de 17-hidroxilasa
La deficiencia de 17-hidroxilasa es rara. Se presenta en la pubertad debido a la producción reducida de andrógenos suprarrenales (hypogonadism)
- Las mujeres con deficiencia de 17-hidroxilasa presentan pubertad tardía: no menstrúan y los senos y el vello púbico no se desarrollan.
- Los machos se presentan con genitales externos ambiguos o parecen ser mujeres (pseudohermafroditismo masculino).
La deficiencia de 17-hidroxilasa provoca una disminución de la producción de cortisol y un aumento de los mineralocorticoides. Las pruebas revelan:
- Hipertensión (hypertension)
- Bajo en potasio
- Renina baja en plasma
- Bajo nivel de cetosteroides en orina 17
- Gonadotropina urinaria alta
El tratamiento es con dexametasona para corregir la hipertensión y testosterona para acelerar la maduración sexual.
Deficiencia de 3-beta-hidroxiesteroide deshidrogenasa
La deficiencia de 3β-hidroxiesteroide deshidrogenasa reduce la producción de todas las hormonas esteroides suprarrenales (cortisol, aldosterona, andrógenos y estrogens) Se presenta en la primera infancia con vómitos, pérdida de sal y ambigüedad genital. Las pruebas revelan:
- Bajo en sodio
- Alto potasio
- DHEA alta en orina
- Cortisol urinario bajo metabolites (a saber, 17 hidroxicorticosteroides).
Conversión de pregnenolona a progesterone está alterado, bloqueando la síntesis de cortisol y aldosterona pero aumentando los andrógenos suprarrenales. En las mujeres, estos andrógenos débiles producen virilización parcial.
La deficiencia de 3β-hidroxiesteroide deshidrogenasa reduce la producción de testosterona en los testículos masculinos para que los genitales de un hombre puedan formarse de manera incompleta.
El tratamiento es el reemplazo de glucocorticoides, fludrocortisona y sex esteroides desde la pubertad en adelante.